






































بررسی انواع سندرم های ژنتیکی و علل ایجاد آنها
تاریخچه ژنتیک و تاثیر آن بر علم پزشکی
ارائه حقایق تاریخی حداقل به اندازه پیگیری واقعیت های علمی چالش برانگیز بوده است.
تاریخچه ژنتیک پزشکی پر از یافته های مهیجی است که به بیماران و خانواده هایشان منفعت بسیاری رسانده است. اما در آینده موفقیت این رشته، با پیشرفت های آتی در تبدیل یافته ها به درمان و پیشگیری از بیماری ها ارزیابی خواهد شد و ما این امتیاز ویژه را داریم تا شاهد چنین پیشرفت هایی در آغاز دوره ای باشیم که نویدبخش عصری پرهیجان و شگرف است.
همچنان که پیشرفت میکنیم نباید فراموش شود پیشینیان ما با وجود کمبود امکانات، با عزم راسخ با آنچه که گاهی به صورت غیر مترقبه به دست آورده اند، پایه های این علم پویا را بنا نهاده اند. پیشرفت های علم ژنتیک طی قرن بیستم واقعا چشمگیر بوده اند. در سال ۱۹۰۰ که اصول مندل منتظر کشف مجدد بودند، کروموزوم ها در آن زمان به سختی قابل مشاهده بوده و علم ژنتیک مولکولی وجود نداشت.
ژنتیک با اکثر رشته های پزشکی مرتبط بوده و از اهمیت ویژه ای برخوردار است. کشف های اخیر نه تنها بر روی اختلالات ژنتیکی و بیماری های نادر ژنتیکی و انواع سندرم های ژنتیکی تاثیر داشته است، بلکه بر بسیاری از بیماری های شایع در بزرگسالان که تنوع ژنتیکی در ایجاد آنها سهیم است مانند بیماری های قلبی – عروقی، بیماری های روحی-روانی و سرطان، اثرات اشاره نشده بر چاقی، عملکردهای ورزشی، توانایی موسیقی، طول عمر و گروهی از تغییرات فیزیولوژیکی و آستانه تحمل دارو نیز موثر بوده اند.
دقیقا مشخص نیست که چه زمانی انسان امروزی (Homo sapiens) در کره زمین ظاهر شد، اما برطبق توافق علمی فعلی و بر اساس یافته های حاصل از فسیل های استخوان های انسانی در اتیوپی، بشر در اطراف آفریقای شرقی در حدود ۲۰۰،۰۰۰ سال پیش می زیسته است. منطقی است که فرض کنیم اجداد اولیه ما به اندازه ما دربارهی موضوع توارث کنجکاو بوده اند و همانند امروز تولد فرزندانی با همه حالت های نقائص فیزیکی را تجربه کردهاند.
با توسعه یک تکنیک مطمئن برای آنالیز کروموزومی در سال ۱۹۵۶ مشخص شد که چندین بیماری به دلیل غیر طبیعی بودن تعداد کروموزومها ایجاد میشوند. طی سه سال علت سندرم داون، سندرم کلاین فلتر و سندرم ترنر مشخص شد. کمی پس از آن سایر سندرمهای تریزومی آتوزومی و اختلالات ژنتیکی نیز شناسائی شدند و طی سال های بعدی بسیاری از سندرمهای بدشکلی چندگانه نیز توصیف شدند که در آنها افزایش یا کاهش ماده کروموزومی وجود داشت.
تاکنون ده ها هزار ناهنجاری کروموزومی در شبکه اطلاعاتی پزشکی به ثبت رسیدهاند و با ظهور تکنولوژی هیبریدسازی ژنومی مقایسهای ریزآرایه (CGH) دو تخصص ژنتیک مولکولی و سیتوژنتیک با هم ادغام شدهاند.

مروری بر تاریخچه ژنتیک پزشکی
- در قرن هفدهم لیون هوک وجود اسپرم و تخمک را شناسایی کرد.
- شکوفایی انقلاب علمی در قرن 18 و 19 موجب جلب توجه دانشمندان و پزشکان به توارث شد که در بین آنها نام دو نفر برجسته تر می باشد. پیر دی مو پرتوئیس (Pierre de Maupertuis) طبیعی دان فرانسوی که صفات توارثی مثل انگشتان اضافی (پلی داکتیلی) و فقدان رنگدانه (آلبینیسم) را بررسی می کرد و با مطالعات شجره نامهها نشان داد که این دو بیماری به روش های متفاوتی به ارث میرسند. جوزف آدامز (Josef Adams) یک پزشک انگلیسی مکانیسم های متفاوت توارثی را شناسایی کرد و مقاله ای در مورد «ویژگی های توارثی فرضی بیماری ها» را منتشر کرد. که هدف آن فراهم آوردن اصول مشاوره ژنتیک در آن زمان بود.
- همچنین اشاره به نام پزشک انگلیسی ادوارد مریون (Edward Meryon) شایان توجه است. این دانشمند اولین فردی بود که مطالعات سیستمیک پاتولوژی-بالینی را در سال 1851 برای سه پسر مبتلا به ناهنجاری های عضلانی مطرح کرد.
- عصر علمی نوین با فعالیت های راهب اتریشی گرگول مندل (Gregor Mendel) آغاز شده که در سال 1865 نتایج آزمایش آمیزش های خالص سازی در باغ نخود فرنگی اش به نام « Natural History Society of Brunn» در بوهمیا ارائه کرد. کمی پس از آن مشاهدات مندل توسط آن انجمن در مجله تعاملات اجتماعی (Transaction of the Society) منتشر شد. سپس تا سال 1900 مورد توجه قرار نگرفت و 16 سال پس از مرگ او برای اولین بار اهمیت کارش مشخص شد. در اصل کارهای مندل را می توان به عنوان کشف ژن ها و اینکه چگونه به ارث می سند، در نظر گرفت.
- اصطلاح ژن اولین بار در سال ۱۹۰۹ توسط گیاه شناس دانمارکی به نام یانسن (Johannsen) به کار رفت که از کلمه پانژن (Pangen) ارائه شده توسط دی وریس اقتباس شده است که در سال ۱۸۶۸ توسط داروین ابداع شد. به منظور قدردانی از کارهای بنیادی مندل، اکنون اصطلاح مندلی بخشی از مجموعه لغات علمی است که برای الگوهای توارث متفاوت با ویژگی تکژنی و نیز برای بیماریهائی ناشی از نقائص تک ژنی به کار میرود.
- والتر ساتن و تئودور بووری اعلام کردند کروموزوم ها میتوانند حاملین توارث باشند.
- توماس مورگان تئوری کروموزومی ساتن را به تئوری ژن تبدیل کرد.
- آلفونز جانسون، مشاهده تشکیل کیاسما بین کروموزوم های همولوگ در میوز
- سیریل دارلینگتون آشکارسازی نقل و انتقالات کروموزومی
- تیجو و لوان تعداد صحیح کروموزوم ها را در سال 1956 مطرح کردند.
- فرد گریفیث، اصل ترانسفورم کنندگی
- اوسلوالد اوری، مکلین مک کارتی، کولین مک لود، تعیین کردند که DNA به عنوان ماده ژنتیکی است.
- فرانسیس کریک، پل زامکنیک، ماهلون هوگلند، مشخص کردن tRNA
- جان دالتون، مشاهده کوررنگی(دالتونیسم) و هموفیلی
- ویکتور مک یوسیک، کاتالوگ بیماری های تک ژنی (OMIM)
انواع سندرم های ژنتیکی
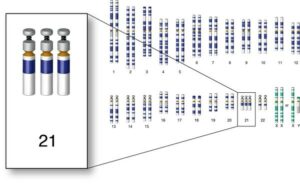
سندرم داون (تریزومی ۲۱)
این بیماری به نام دکتر لانگدون داون(Langdon down) که اولین بار بیماری را در گزارشات سخنرانی بالینی در بیمارستان لندن در سال ۱۸۶۶ توصیف کرد، نامگذاری شده است. اساس کروموزومی سندرم داون مشخص نبود تا اینکه در سال ۱۹۵۹ توسط لژون (Lrjeune) و همکارانش در پاریس معین گردید.
سندرم دان یا نشانَگان دان ( Down syndrome) یک بیماری ژنتیکی است که به دلیل حضور تمام یا بخشی از یک کروموزوم اضافی در جفت کروموزوم ۲۱ به وجود میآید که در اصطلاح علمی تریزومی ۲۱ نیز نامیده میشود.
تریزومی ۲۱، کروموزوم اضافی در بیش از ۹۰ درصد موارد منشا مادری دارد در واقع یک همراهی قوی بین میزان بروز سندروم داون با افزایش سن مادر وجود دارد و مطالعات DNA نشان دادهاند که معمولا در نتیجه عدم تفکیک صحیح کروموزومی در میوز I مادری ایجاد می شود. و همچنین جابه جایی روبرت سونین مسئول ۴ درصد کل موارد بوده و به طور تقریبی یک سوم والدین ناقل این جابه جایی هستند.
میزان بروز کلی تولدها وقتی مطابق با اثر گسترده برنامههای غربالگری پیش از تولد باشد تقریبا ۱ به ۱۰۰۰ در انگلستان میباشد. در ایالات متحده میزان بروز در زمان تولد حدود ۱ به ۸۰۰ تخمین زده شده است.
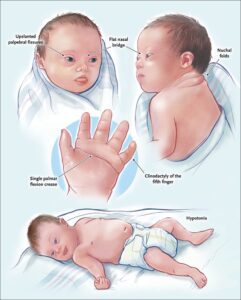
علائم بالینی
شایعترین مشخصه سندرم داون هنگام تولد هیپنوتونی شدید (Hypotonaia) است.(هیپوتونی حالتی از کاهش قدرت و یا کشش عضلات است که میتواند در هر ساختاری از بدن مشاهده شود.) معمولا علائم چهرهای همراه با شکاف پلکی با شیب رو به بالا، گوش های کوچک و زبان بزرگ تشخیص سریع سندرم را تسریع میبخشند. کودکان مبتلا طیف وسیعی از توانائیهای ذهنی را با مقادیر متفاوت IQ از ۲۵ تا ۷۵ را نشان میدهند. IQ متوسط بالغین جوان در حدود ۴۰ تا ۴۵ میباشد.
این بیماری دارای علائم مختلف از جمله ناهنجاری های عمده یا خفیف در ساختار یا عملکرد ارگان ها میباشد. از جمله علائم عمده و زودرس که تقریباً در همه بیماران مشاهده میشود وجود مشکلات یادگیری و نیز محدودیت و تأخیر رشد و نمو میباشد.
در حال حاضر هیچ راهی برای پیشگیری از این بیماری وجود ندارد. بنابراین افرادی که در سنین بالاتر باردار میشوند یا سابقهی بیماریهای ژنتیکی مثل سندروم داون در خانواده آن ها وجود دارد، باید پیش از بارداری از پزشک متخصص مشاوره بگیرند.
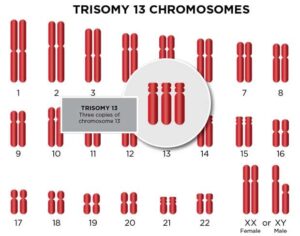
سندرم پاتائو ( ترزومی 13)
سندرم پاتو یک ناهنجاری کروموزومی است که در آن برخی یا همه سلولهای بدن حاوی مواد ژنتیکی اضافی از کروموزوم 13 هستند. سندرم پاتائو نخستین بار در سال 1960 توصیف شد. این سندرم می تواند به این دلیل رخ دهد که هر سلول حاوی یک نسخه کامل اضافی از کروموزوم 13 یا هر سلول حاوی یک نسخه جزئی اضافی از کروموزوم 13 و یا به دلیل وجود دو کروموزوم متفاوت رخ دهد. خطوط سلولی – یکی سالم با تعداد صحیح کروموزوم 13 و دیگری که حاوی یک نسخه اضافی از سندرم پاتاو کروموزوم موزاییک است. تریزومی 13 کامل به دلیل عدم تفکیک کروموزوم ها در طول میوز ایجاد می شود (شکل موزاییک ناشی از عدم جدایی در طول میتوز است).
علائم بالینی
این سندرم با ناتوانی شدید ذهنی و ناهنجاری های فیزیکی در بسیاری از قسمت های بدن همراه است. ناهنجاری های قلبی حداقل در 90 درصد موارد رخ می دهد. افراد مبتلا به تریزومی 13 اغلب دارای نقایص قلبی، ناهنجاری های مغزی یا نخاعی، چشم های بسیار کوچک یا ضعیف (میکروفتالمی)، انگشتان اضافی، سوراخ در لب (شکاف لب) با یا بدون سوراخ در سقف دهان، شکاف کام و هیپوتونی هستند.
به دلیل وجود چندین مشکل پزشکی تهدید کننده زندگی، بسیاری از نوزادان مبتلا به تریزومی 13 در اولین روزها یا هفته های زندگی خود می میرند. تنها 5 تا 10 درصد از کودکان مبتلا به این بیماری از سال اول زندگی خود می گذرند. در موارد نامعمول که زمان طولانی تری پس از تولد زنده بمانند، همراه با مشکلات شدید یادگیری خواهند بود.
بروز این سندرم همراه با افزایش سن مادر، شایعتر می شود در نتیجه کروموزوم اضافی منشاء مادری دارد. تقریبا 10 درصد موارد به صورت موزائیسم همراه با نوآرایی های نامتعادل، به خصوص جابه جایی روبرت سونین در سندرم پاتائو می باشد.
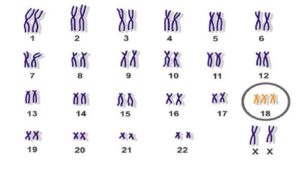
سندرم ادوارد ( تریزومی 18)
این بیماری اولین بار در سال 1960 توصیف شد. بسیاری از علائم سندرم ادوارد و پاتائو مشترک می باشند. این نام به افتخار ژنتیکدان انگلیسی جان هیلتون گرفته شده است. میزان بروز هر دو بیماری تقریبا 1:5000 است و اکثر نوزادان در طی اولین روزها یا هفته های زندگی می میرند.
علائم بالینی
سندرم ادوارد یک اختلال ژنتیکی است که به دلیل وجود یک نسخه کامل یا بخش اضافی از کروموزوم 18 ایجاد می شود. بسیاری از اعضای بدن تحت تأثیر قرار می گیرند. نوزادان اغلب کوچک به دنیا می آیند و دارای نقایص قلبی هستند. ویژگی های دیگر عبارتند از: سر کوچک، آرواره کوچک، مشت های گره کرده با انگشتان روی هم، و ناتوانی شدید ذهنی.
بروز این ناهنجاری همراه با افزایش سن مادر، شایعتر است و کروموزوم اضافی منشاء مادری دارد. گاهی اوقات، همه سلول ها دارای کروموزوم اضافی نیستند که این مورد به عنوان تریزومی موزاییک شناخته می شود، و علائم در این مورد ممکن است شدیدتر باشد. سونوگرافی در دوران بارداری شک به این بیماری را افزایش می دهد که می تواند با آمنیوسنتز تایید شود.
پس از داشتن یک فرزند مبتلا به این عارضه، خطر داشتن فرزند دوم معمولاً حدود یک درصد است.این دومین بیماری شایع ناشی از کروموزوم اضافی در بدو تولد، پس از سندرم داون است. برخی از مطالعات نشان می دهد که تعداد بیشتری از نوزادانی که تا زمان تولد زنده می مانند، دختر هستند. بسیاری از مبتلایان پیش از تولد می میرند. در مواردی که زمان طولانی تری پس از تولد زنده می مانند، بیش از یک سال زندگی حدود 5 تا 10 درصد است.
سندرم پرادرویلی و سندرم آنجلمن
این دو بیماری جایگاه ویژه ای در ژنتیک به عنوان الگوهایی برای نشان گذاری ژنومی دارند. این بیماری به افتخار پزشکان سوئیسی آندره آ پرادر و هاینریش ویلی که به همراه الکسیس لابهارت آن را به تفصیل در سال 1956 توصیف کردند، نامگذاری شده است سندرم پرادرویلی (PWS) تقریبا با فراوانی 1 در 20000 تولد رخ می دهد.
علائم بالینی
وجود این بیماری با قد کوتاه، چاقی، هیپوگنادیسم و مشکلات یادگیری مشخص می شود. دارای حالت بسیار شل (هیپوتونیک) و همراه با تغذیه کم در نوزادی می باشند اما بعدا دچار پرخوری و چاقی همراه با مشکلات یادگیری خفیف تا متوسط می شوند. افراد مبتلا به پرادرویلی(PWS) دارای یک حذف بینابینی حدودا 2Mb در بخش پروکسیمال بازوی بلندکروموزوم 15 در موقعیت 15q11-q13 می باشند، که با روش های سیتوژنتیکی مرسوم قابل روئیت بوده و 15 درصد از موارد حذف تحت میکروسکوپی دیگر را می توان با روش هیبریدسازی فلورسنت درجا (FISH) یا روش های مولکولی مشخص نمود.
آنالیز DNA نشان داده که کروموزوم حذف شده تقریبا همیشه همولوگ به ارث رسیده پدری است. 25 درصد تا 30 درصد بقیه مواردمبتلا به PWS که حذف کروموزومی ندارند، دارای دیزومی تک والدی مادری می باشند. از لحاظ عملکردی این حالت متعادل حذف در کروموزوم 15 به ارث رسیده پدری است.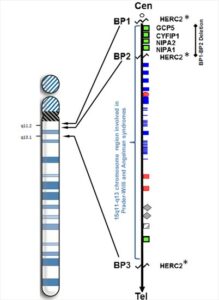
سندرم آنجلمن (AS) تقریبا با فراوانی 1 در 15000 تولد رخ می دهد و همراه با صرع، مشکلات شدید یادگیری، قدم های آتاکسیک یا نامتوازن و چهره ای خندان مشخص می شود و مشکلات شدید یادگیری دارند.
تقریبا 70 درصد افراد مبتلا به سندرم AS دارای یک حذف بینابینی در همان ناحیه 15q11-q13 دخیل در PWS، اما در این مورد در همولوگ به ارث رسیده مادری می باشند. در 5 درصد دیگر موارد مبتلایان AS، در اثر دیزومی تک والدی پدری ایجاد شده است. برخلاف سندرم پرادرویلی علائم سندرم آنجلمن توسط حذف یک ژن UBE3A می باشد.
در بسیاری از آزمایشگاه های تشخیصی ژنتیکی، یک آزمایش ساده DNA برای تشخیص هر دو بیماری PWS و AS بر پایه ویژگی های متفاوت متیلاسیون DNA در لوکوس 15q11-q13 به کار می رود.
سندرم بک ویت ویدمن(Bechwith- Wiedemann Syndrome)
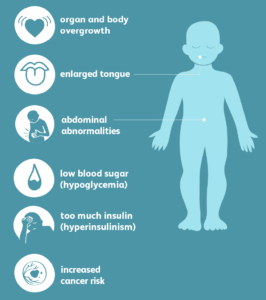
سندرم بک ویت ویدمن(BWS) یک بیماری هتروژن از لحاظ بالینی است که ویژگی اصلی آن رشد بیش از حد می باشد. این سندرم اولین بار در سال های 1963 و 1964 توصیف شدکه ویژگی های عمده آن بزرگی بدن ( رشد زیاد قبل یا بعد از تولد). BWS در اثر تغییرات کروموزوم 11p15.5 ایجاد می شود
علائم بالینی
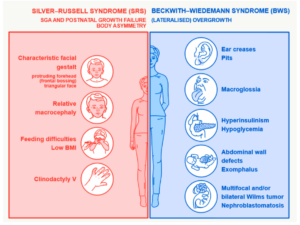
شایع ترین اختلال، رشد بیش از حد و استعداد ابتلا به سرطان است و با طیف وسیعی از علائم و یافته های فیزیکی مشخص می شود که در محدوده و شدت از فردی به فرد دیگر متفاوت است. ماکروگلوسیا (بزرگی زبان)، نقائیص دیواره شکمی، فتق نافی و از هم فاصله گرفتن ماهیچه های راست شکمی به موازات محور میانی دیواره شکم و هیپوگلیسمی و … از دیگر علائم این سندرم می باشند.
سندرم راسل سیلور (Russell Silver Syndrome)
دکتر هنری سیلور ویژگیهای خاصی از سندرم راسل-سیلور را در سال 1953 کشف کرد. دکتر الکساندر راسل در سال 1954 ویژگیهای دیگری از این بیماری را کشف کرد. برای تقریباً 20 سال، محققان فکر میکردند سیلور و راسل دو وضعیت جداگانه پیدا کردهاند. بعداً مشخص شد که پزشکان ویژگیهای متفاوتی از یک بیماری را مشاهده میکنند. در ایالات متحده، این بیماری معمولاً سندرم راسل-سیلور، ولی در اروپا، این بیماری سندرم سیلور-راسل نامیده می شود.
سندرم راسل-سیلور یک بیماری ژنتیکی نادر است که از هر 15000 تا 100000 تولد یک مورد را تحت تاثیر قرار می دهد. فراوانی دقیق این وضعیت ناشناخته است. ویژگی های سندرم راسل-سیلور اغلب شبیه سایر اختلالات رشد و شرایط مادرزادی است. به همین دلیل، بسیاری از موارد سندرم راسل-سیلور ممکن است تشخیص داده نشود. حدود 10 درصد موارد به دلیل دیزومی تک والدی مادری ایجاد شده که نمایانگر این است که کروموزوم مربوطه نشان گذاری می شود.
بر خلاف مضاعف سازی های پدری 11p15 که باعث رشد بیش از حد و BWS می شود، مضاعف سازی های مادری این ناحیه با تاخیر رشد همراه است. اخیرا نشان داده شده است که در حدود یک سوم موارد سندرم راسل-سیلور (RSS) به دلیل ناهنجاری های نشان گذاری در لوکوس 11p15.5 می باشد.
علائم بالینی
این بیماری به خوبی شناخته شده است و دارای ویژگی های مخالف BWS بوده و در واقع با تاخیر رشد قبل و بعد از تولد مشخص می گردد. محیط پیرامون سر نسبتا طبیعی، صورت کمی کوچک و مثلثی شکل که ایجاد یک ظاهر هیدروسفالیک کاذب می کندو ممکن است بدن نامتقارن باشد. علائم دیگر شامل وزن کم هنگام تولد و چندین ناهنجاری فیزیکی است.
تصور می شود که این عارضه ناشی از تغییرات در ژن های خاصی است که رشد را کنترل می کنند. تشخیص را می توان از طریق معاینه بالینی و آزمایش ژنتیک انجام داد. چشم انداز سندرم راسل-سیلور مثبت است. سندرم راسل-سیلور می تواند هر کودکی را درگیر کند. این اختلال بر پسران و دختران به طور مساوی تأثیر می گذارد.
سندرم میلر-دیکر (Miller-Dieker Syndrome)
نام “سندرم میلر-دیکر” از پزشکانی به نام جیمز کیو میلر و اچ دیکر گرفته شده است که اولین بار این بیماری را در دهه 1960 توصیف کردند. سندرم میلر-دیکر یک اختلال ژنتیکی نادر است که باعث صاف شدن قسمت بیرونی مغز کودک (قشر مغز) می شود. مغز چین و چروکها و شیارهای پیچیده معمولی را ندارد.
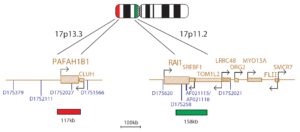
این سندرم در اثر ریز حذف کروموزومی در ناحیه 17p13.3، واقع در انتهای دیستال بازوی کوچک کروموزوم 17 ایجاد می شود. کودکان مبتلا به سندرم میلر-دیکر بخشی از کروموزوم 17 را از دست داده اند. این از دست دادن یک یا چند ژن به طور تصادفی و بدون هیچ دلیل شناخته شده ای اتفاق می افتد. حذف ژن ممکن است در اسپرم، تخمک یا پس از لقاح رخ دهد. میزان شیوع این سندرم نامشخص است.
علائم بالینی
شامل لیسنسفالی (Lissencephaly) بوده که در آن سطح مغز صاف و بدون شیار (gyri) است. مغز کوچک (Microcephaly) و پیشانی بلند از علائم دیگر آن است. مرگ در اوایل زندگی رخ می دهد.
ریز حذف کروموزومی در ناحیه 17p13.3 در اکثر موارد (حدود 90%) بیماران مبتلا به سندرم میلر دیکر با استفاده از سیتوژنتیک و روش های ملکولی قابل تشخیص است. اما این ریز حذف فقط در 15 درصد بیماران مبتلا به لیسنسفالی ایزوله (ابتلا لیسنسفالی بدون وجود علائم چهره و دیگر علائم) قابل تشخیص است.
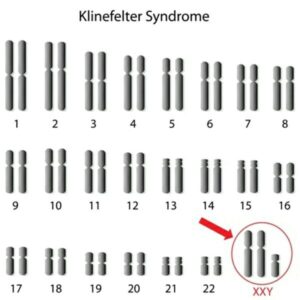
سندرم کلاین فلتر (Klinefelter syndrome)
نخستین بار در سال 1942 شرح داده شد. این بیماری نسبتا شایع با میزان بروز 1:1000 در تولدهای زنده مردان در سال 1959 مشخص شد که به دلیل وجود یک کروموزوم X اضافی ایجاد می شود. معمولا در کاریوتایپ یک کروموزوم اضافی مشخص می شود. مطالعات مولکولی نشان داده اند که احتمال تقریبا یکسانی برای توارث این بیماری از پدر و مادر وجود دارد. مواردی که از طرف مادر ایجاد می شوند، با افزایش سن مادر مرتبط اند. بخش کمی نیز به دلیل موزائیسم نشان می دهند. کروموزوم X (مثلا 48XXXY یا 49XXXXY ) مشاهده شده اند.
علائم بالینی
بیماران در کودکی ممکن است اختلالات حرکتی همراه با مشکلات یادگیری خفیف به خصوص در ارتباط با مهارت های گفتاری را نشان دهند. بالغین کمی قد بلندتر از متوسط افراد، همراه با پاهای بلند می باشند. تقریبا 30% ژنیکوماستی نسبتا شدید را نشان می دهند، همه آنها ناباروند زیرا فاقد اسپرم در مایع منی شان بوده (آزواسپرمی) و بیضه های کوچک نرم دارند. با استفاده از تکنیک های آسپریلاسیون اسپرم از بیضه و تزریق اسپرم داخل سیتوپلاسمی در تعداد کمی از مبتلایان انجام شده و باروری حاصل می شود. درمان با تستوسترون نیز پس از بلوغ برای ایجاد صفات ثانویه جنسی و جلوگیری بلندمدت از پوکی استخوان سودمند می باشد.
سندرم بلوم ( Bloom Syndrome)
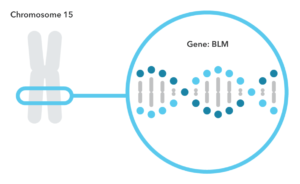
سندرم بلوم یک اختلال ژنتیکی اتوزومال مغلوب نادر است که با کوتاهی قد، مستعد ابتلا به سرطان و بی ثباتی ژنومی مشخص می شود. این بیماری برای اولین بار توسط دکتر دیوید بلوم، متخصص پوست در سال 1954 کشف و توصیف شد. به طور طبیعی ژن سندرم بلوم نقش عمده ای در حفظ پایداری ژنوم ایفا می کند. اگر در حالت هموزیگوت معیوب باشد، آنگاه ترمیم DNA نیز مختل بوده و میزان نوترکیبی بین کروماتیدهای خواهری به طور چشمگیری افزایش می یابد. این مورد را می توان با بررسی تبادلات کروماتیدهای خواهری مشاهده کرد. این سندرم در اثر جهش در ژن BLM که عضوی از خانواده هلیکاز DNA RecQ است ایجاد می شود.
علائم بالینی
کودکان مبتلا به این بیماری با توارث آتوزوم مغلوب، جثه ای کوچک همراه با لکه های پوستی حساس به نور در صورت و سطوح کاهش یافته ایمنوگلوبولینی (IgM و IgA) دارند. سلول های کشت شده افزایش فراوانی شکستگی های کروموزومی را نشان می دهند، ژن مربوط به سندرم بلوم بر روی کروموزوم 15q26 نقشه برداری شده است و کد کننده یک عضو از آنزیم های DNA هلیکاز ها است.
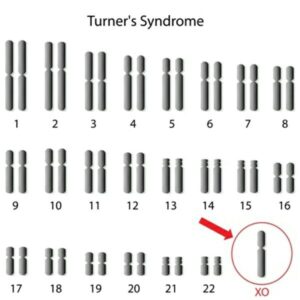
سندرم ترنر ( 45X)
این بیماری اولین بار در سال 1938 توصیف شد. فقدان جسم بار همراه با حضور فقط یک کروموزوم X در سال 1954 مشاهده شد و تایید سیتوژنتیکی آن در سال 1959 فراهم شد. اگرچه در لقاح و سقط های خودبه خودی شایع می باشند. میزان بروز نوزادان دختر زنده به دنیا آمده کم است و به طور تخمینی از 1:5000 تا 1:10000 می باشد.
شایع ترین یافته کروموزومی 45X است. 80% موارد در اثر حذف یک کروموزوم جنسی (X یا Y) در میوزپدری ایجاد می شود. در بخش قابل توجهی از موارد، موزائیسم کروموزومی وجود دارد و افرادی که یک رده سلولی طبیعی (46XX) دارند دارای شانس باروری می باشند.
علائم بالینی
علائم در هر زمانی از لقاح تا زندگی بزرگسالی مشاهده می شود. سندرم ترنر طی سه ماهه دوم حاملگی در نتیجه یک اولتراسونوگرافی معمول تشخیص داده می شود که همراه با اِدم عمومی یا تورم ناحیه گردنی می باشد. در زمان تولد بسیاری از کودکان مبتلا به سندرم ترنر کاملا طبیعی به نظر می رسند.
سایر یافته ها ممکن است شامل خط رویش موی پایین، تنگی آئورت و … می باشد. ضریب هوشی در سندرم ترنر طبیعی است. با این حال مطالعات نشان داده که برخی تفاوت ها در آگاهی های اجتماعی و سایر مهارت های عملکردی در سطوح بالاتر وابسته به اینکه کروموزوم X آنها منشا پدری یا مادری داشته باشد مشاهده می شود.
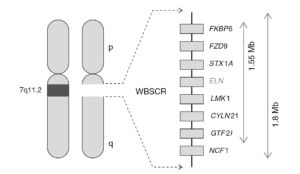
سندرم ویلیامز (Williams syndrome)
سندرم ویلیامز اولین بار توسط ویلیامز در سال 1961 گزارش شد و سپس توسط بورن بیشتر مشخص گردیدبنابراین گاهی به نام سندرم ولیامز-بورن شناخته می شود. سندرم ویلیامز به دلیل ریز حذف کروموزوم 7q11.2 رخ می دهد و تشخیص این بیماری توسط روش FISH تایید می گردد.
علائم بالینی
سندرم ویلیامز یک اختلال رشدی است که بسیاری از قسمت های بدن را تحت تاثیر قرار می دهد. این وضعیت با ناتوانی ذهنی یا مشکلات یادگیری خفیف تا متوسط، ویژگی های شخصیتی منحصر به فرد، ویژگی های متمایز صورت و مشکلات قلبی و عروقی (قلبی-عروقی) مشخص می شود. کودکان خردسال مبتلا به سندرم ویلیامز دارای ویژگی های متمایز صورت از جمله پیشانی پهن، پف دور چشمها، پل صاف بینی، گونه های پر و چانه کوچک هستند. بسیاری از افراد مبتلا مشکلات دندانی مانند دندان های کوچک، فاصله زیاد، کج بودن یا از دست دادن دندانها دارند. کودکان بزرگتر و بزرگسالان معمولاً صورت درازتر با دهان پهن و لب های پر دارند.
سندرم CHARGE
تشخیص این سندرم برمبنای مجموعه از ویژگی های بالینی خاص است. نام “CHARGE” روشی هوشمندانه (در سال 1981) برای اشاره به مجموعه ای از ویژگی های تازه شناخته شده بود که در تعدادی از کودکان دیده می شود. در طول سال ها، مشخص شده است که CHARGE در واقع یک سندرم است و حداقل یک ژن ایجاد کننده آن کشف شده است.
سندرم CHARGE با جهش ژن CHD7 ارتباط دارد. در سال 2004 وجود یک ریزحذف به طول 2/3 کیلو باز در 8q12 در دو نفر از افراد به صورت حذف de novo گزارش شد. آنالیز تعیین توالی ژن های موجود در این ناحیه باعث کشف جهش در ژن CHD7 در 10 نفر از 17 نفر مبتلا به سندرم CHARGE شد که فاقد ریز حذف 8q12 بودند.
شیوع بیماری 1 در 8500 تا 10000 است. چون در اکثر موارد جهش عامل بیماری به صورت de novo رخ می دهد، خطر عود مجدد برابر جمعیت عادی، حدود 0.1 درصد است. امید به زندگی مبتلایان بالای 5 سال حدود 70 درصد است و اکثر موارد مرگ و میر مربوط به این سندرم در سال اول زندگی رخ می دهد.
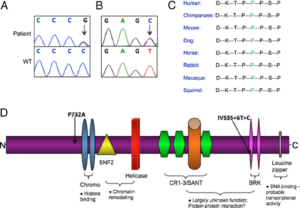
علائم بالینی
در افراد دارای جهش در ژن CHD7، ناهنجاری های قلبی، کلوبوم چشم و عدم تقارن صورت بیشتر مشاهده شد. ارتباط ژنوتیپ-فنوفیپ خاصی بر مبنای این جهش ها مشخص نشد و شدت بیماری ارتباطی با طول پروتئین CHD7 جهش یافته نداشت.
واژه CHARGE برگرفته از علائم بالینی ذیل است:
1- C برگرفته از کلوبومای چشم (Coloboma)
2- H برگرفته از بیماری قلبی (Heart)
3- A برگرفته از انسداد (Atreisa)دریچه بین حفره بینی و نازوفارنکس
4- R برگرفته از عقب ماندگی (Retarded) رشد وتکوین یا ناهنجاری های سیستم اعصاب مرکزی
5- G برگرفته از هیپوپلازی مجاری تناسلی (Genital)
6- E برگرفته از ناهنجاری های گوش و یا ناشنوایی (Ear)
علائم بارز دیگر شامل فلج صورت، چانه کوچک، شکاف، کام، مشکلات بلع و فیستول بین (مجاری غیر طبیعی) نای و مری و اودیوگرام مثلثی شکل هستند.
سندرم پالیستر – کیلیان (Pallister – Killian)
سندرم پالیستر-کیلیان (تترازومی 12p) یک ناهنجاری نادر کروموزومی است که به دلیل داشتن یک کروموزوم اضافی ایجاد می شود. ارثی نیست و به طور خود به خود در کودک اتفاق می افتد. تمام موارد ثبت شده تا به امروز پراکنده بوده است.
در این اختلال افراد دارای چندین ویژگی دیسمورفیک و عقب ماندگی ذهنی شدید می باشند. این بیماری اگرچه بسیار نادر است، اما از این جهت اهمیت دارد که در آن یک نوع موزاییسم مختص بافت دیده می شود. زیرا کروموزوم اضافه فقط در بافت های خاصی دیده می شود.
سندرم پالیستر-کیلیان معمولاً به دلیل وجود یک کروموزوم اضافی غیر طبیعی 12 به نام ایزوکروموزوم 12p ایجاد می شود. سایر تغییرات کروموزومی پیچیده تر که شامل کروموزوم 12 می شود ممکن است در موارد نادر باعث ایجاد سندرم شود. این کروموزوم اضافی هم از طریق پدر و هم مادر به ارث می رسد، اگرچه از 6 گزارش منتشر شده، در 5 مورد منشا کروموزوم اضافی مادری بوده است.
علائم بالینی
علائم و نشانههای سندرم موزاییک پالیستر-کیلیان متفاوت است. افراد مبتلا دارای چهره ای خشن همراه با ناهنجاری های رنگدانه ای پوست هستند. یزرگسالان مبتلا نیز داراری عقب ماندگی شدید ذهنی، صرع، صورت بزرگ، پیشانی بزرگ و زبان بزرگ هستند. چند مورد با فنوتیپ خفیف تر بیماری نیزگزارش شده است. نوزادان مبتلا شدیدا شل (هیپوتونیک) هستند.
راه های تشخیص سندرم پالیستر-کیلیان
مطالعه کروموزوم سلول های پوست (فیبروبلاست): پزشکان چندین نواحی پوست را آزمایش می کنند و مواد ژنتیکی را با هم مقایسه می کنند. اگر پزشک مشکوک باشد که فرد به سندرم موزاییک پالیستر-کیلیان مبتلا است، احتمالاً از این آزمایش برای تأیید تشخیص استفاده خواهد کرد. پزشکان آزمایش سلول های پوستی را به جای آزمایش خون ترجیح می دهند، زیرا شرایط موزاییکی را زمانی که همه سلول ها تحت تأثیر قرار نمی گیرند مشخص می کند.
هیبریداسیون درجا فلورسنت (FISH): در طول این آزمایش خون تشخیصی بسیار دقیق، پزشکان از مواد ژنتیکی در سلول های کودک نقشه برداری می کنند. آزمایش FISH به پزشکان اجازه می دهد تا ببینند کدام کروموزوم دارای مواد ژنتیکی اضافی یا مفقود است. از آزمایش Array-CGH برای تایید بیماری پالیستر کیلیان نیز می توان استفاده کرد.
سندرم پالیستر کیلیان را می توان قبل از تولد با آمنیوسنتز یا نمونه برداری از پرزهای کوریونی (CVS) نیز تشخیص داد. این آزمایشها شامل برداشتن مقدار کمی مایع آمنیوتیک که نوزاد را در رحم احاطه کرده است، یا نمونهای از پرزهای کوریونی، بافتی از جفت که دارای سلولهایی از نوزاد است، میشود.
سندرم سوتوس (Sotos Syndrome)
این بیماری اولین بار در سال 1964 به عنوان یک سندرم «رشد بیش از حد» توصیف شد و قبلا به آن ژیگانتیسم مغزی می گفتند. سندرم سوتوس معمولاً به دلیل تغییر ژنتیکی در ژن NSD1 ایجاد می شود و به صورت اتوزومال غالب به ارث می رسد. حدود 95 درصد موارد به دلیل تغییر ژنتیکی جدید در فرد مبتلا بوده و به صورت پراکنده (ارثی نیستند) رخ می دهد.
جهش در ژن NSD1 باعث ایجاد سندرم سوتوس می شود. ژن NSD1 به بدن شما دستورالعمل هایی در مورد چگونگی رشد و نمو می دهد. هنگامی که یک جهش بر ژن NSD1 تأثیر می گذارد، ژن ها نمی توانند رشد بدن را تنظیم کنند و باعث می شود کودکان مبتلا به سندرم سوتوس قد بلندتری نسبت به همسالان خود داشته باشند.
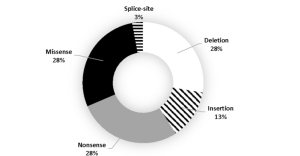
امکان بروز جهش های مختلف بر روی ژن NSD1
علائم بالینی
سندرم سوتوس یک بیماری نادر است که از هر 14000 تولد یک نفر را تحت تاثیر قرار می دهد. اغلب، سندرم سوتوس علائم مشابهی با سایر بیماری ها دارد. به همین دلیل بسیاری از موارد تشخیص داده نمی شوند و تشخیص سندرم سوتوس می تواند چالش برانگیز باشد زیرا علائم مشابهی با سایر بیماری های رایج دارد.
پزشک متخصص تشخیص را با معاینه فیزیکی کودک برای جستجوی علائم این بیماری آغاز می کند. اگر علائم مشکوک به این بیماری وجود داشته باشد، آزمایش ژنتیکی پس از معاینه فیزیکی انجام می شود، پزشک متخصص نمونه کوچکی از خون کودک را برای جستجوی جهش در ژن NSD1 می گیرد. اگر نتایج یک جهش در ژن NSD1 را نشان دهد، پزشک متخصص این بیماری را تشخیص می دهد.
وزن نوزاد در هنگام تولد معمولا بیش از حد نرمال بوده و ماکروسفالی نیز دیده می شود. صورت دارای مشخصات خاصی است که در آن پیشانی بسیار برجسته بوده و هایپرتلوریسم به همراه شکاف پلکی رو به پایین و یک بینی با شکل خاصی در کودکان دیده می شود و همچنین چانه نوک تیز است.
سندرم نونان (Noonan Syndrome)
سندرم نونان یک اختلال ژنتیکی است که به طور معمول در بدو تولد مشهود است (مادرزادی). این اختلال با طیف گسترده ای از علائم و ویژگی های فیزیکی مشخص می شود که از نظر دامنه و شدت بسیار متفاوت است. در ابتدا در سال ۱۹۶۳ توسط Nononan و Ehmk توضیح داده شد، این بیماری که به خوبی شناخته شده دارای میزان بروز ۱ در ۲۰۰۰ است و به طور مساوی در هر دو جنس رخ می دهد.
در اکثر موارد سندرم نونان یک اختلال ژنتیکی اتوزومال غالب است که در اثر ناهنجاری (جهش) در بیش از هشت ژن ایجاد می شود. پنج ژن متداول درگیر عبارتند از: PTPN11 (50%)، SOS1 (10-13%)، RAF1 (5%)، RIT1 (5%) و KRAS (کمتر از 5%). افراد کمتری دارای جهش در NRAS، BRAF، MEK2، RRAS، RASA2، A2ML1 و SOS2 هستند. اختلالات شبه نونان در ارتباط با جهش در SHOC2 و CBL یافت می شود. در درصد کمی از بیماران فاقد جهش PTPN11 مشاهده شده اند.
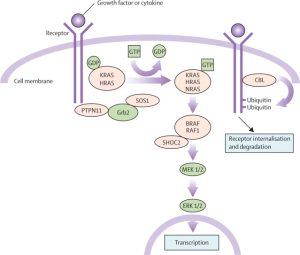
علائم بالینی
نشانه های نونان مشابه علائم موجود در زنان مبتلا به سندرم ترنر است که شامل قامت کوتاه قد، گردن پره دار، زاویه آرنج و بیماری قلبی مادرزادی است. تصور می شد که این بیماران نوعی از سندرم ترنر هستند اما مواردی همانند انسداد ریوی رایج تربن نقص بوده و نقص دیواره دهلیزی، نقص دیواره بطنی و گاهی کاردیومیوپاتی هیپرتروفیک نیز وجود دارد.
در سال ۱۹۹۴ در خانواده ای با سه نسل، سندرم نونان در ناحیه 12q22 نقشه برداری شد. اما در سال ۲۰۰۱ بود که جهش هایی در ژن PTPN11 گزارش شد. نظرها سریعا به ارتباط ژنوتیپ-فنوتیپ معطوف شد.
در برخی موارد، بر اساس نتایج سونوگرافی جنین، که یک تکنیک تصویربرداری تخصصی که در آن از امواج صوتی برای ایجاد تصویری از جنین در حال رشد استفاده می شود، ممکن است قبل از تولد به سندرم نونان مشکوک شوند. تشخیص سندرم نونان ممکن است به دلیل غیرطبیعی بودن غربالگری سه گانه سرم مادر، تشخیص مایع آمنیوتیک بیش از حد جنین در اطراف کیسه آمنیوتیک (پلی هیدرآمنیوس)، وجود تورم غیر طبیعی کیستیک متشکل از عروق لنفاوی متسع در ناحیه گردن در نظر گرفته شود.
سندرم نونان در بدو تولد یا اوایل نوزادی بر اساس ارزیابی بالینی کامل، شناسایی یافتههای فیزیکی مشخصه و انواع آزمایشهای تخصصی تشخیص داده میشود. اگر در دوران بارداری مشکوک به سندرم نونان باشد، آزمایش ژنتیک مولکولی توسط مایع آمنیوتیک یا آنالیز Cell Free DNA در دسترس است. برای تشخیص دقیق تر سندرم نونان از یافته های رادیوگرافی نظیر MRI, CT Scan ، سونوگرافی و اکوکاردیوگرام استفاده می کنند.
سندرم اسمیت مگنیس (Smith-Magenis Syndrome)
سندرم اسمیت-مگنیس (SMS) یک اختلال رشدی است که بسیاری از قسمت های بدن را تحت تاثیر قرار می دهد. ویژگی های اصلی این بیماری شامل ناتوانی یادگیری خفیف تا شدید، ویژگی های متمایز صورت، اختلالات خواب و مشکلات رفتاری است. سندرم اسمیت-مگنیس از هر 25000 نفر یک نفر را تحت تاثیر قرار می دهد. این سندرم به دلیل ناهنجاری در بازوی کوتاه (p) کروموزوم 17 است و گاهی اوقات سندرم 17p نامیده می شود.
این سندرم ریز حذف به دلیل حذف کروموزومی 17p11.2 که اغلب از لحاظ سیتوژنتیکی قابل مشاهده است، ایجاد می شود. همانند سندرم دی جورج مکانیسم حذف در بسیاری از موارد شامل ترکیبی همولوگ بین توالی های LCR مجاور می باشد.

علائم بالینی
ویژگی های فیزیکی خیلی مشخص نمی باشند، اما مشکلات قلبی در یک سوم موارد، اسکولیوز در اوخر کودکی در بیش از نیمی از موارد و نقص شنوائی در حدود دو سوم موارد مشاهده می شود. مبتلایان به سندرم اسمیت مگنیس معمولاً دارای شخصیت های با محبت و جذاب هستند، اما اکثر آنها مشکلات رفتاری نیز دارند.
عصبانیت مکرر، پرخاشگری، اضطراب، آسیب به خود، از جمله گاز گرفتن، ضربه زدن به سر و برداشتن پوست بسیار رایج است. در آغوش گرفتن مکرر خود یک ویژگی رفتاری است که ممکن است منحصر به سندرم اسمیت مگنیس باشد. برخی از افراد مبتلا به این عارضه نیز به طور اجباری انگشتان خود را لیس می زنند و صفحات کتاب و مجلات را ورق می زنند (رفتاری که به عنوان “لیس و تلنگر” شناخته می شود).
سایر علائم و نشانه های سندرم اسمیت ماگنیس شامل کوتاهی قد، انحنای غیرطبیعی ستون فقرات (اسکولیوز)، کاهش حساسیت به درد و دما و صدای خشن است. برخی از افراد مبتلا به این اختلال ناهنجاری های گوش دارند که منجر به کاهش شنوایی می شود. افراد مبتلا ممکن است ناهنجاری های چشمی نیز داشته باشند که باعث نزدیک بینی (نزدیک بینی) و سایر مشکلات بینایی می شود، اگرچه کمتر شایع است، نقایص قلبی و کلیوی نیز در افراد مبتلا به سندرم اسمیت-مگنیس گزارش شده است.
تشخیص سندرم اسمیت مگنیس با استفاده از تکنیک هیبریداسیون در جای فلورسنت (FISH) و G-banding در آزمایشگاه ژنتیک بررسی می شد. اما امروزه از تکنیک MLPA و real-time PCR که سریع و بازده بالاتری دارند استفاده می شود.با اینکه که تعداد موارد بالینی SMS تشخیص داده شده توسط تکنیک های مولکولی در حال افزایش است، اما گروهی از افراد با ویژگی های SMS و بدون حذف 11p11.2 یا جهش در RAI1 نیز در حال ظهور هستند.
